Abstract
Abstract
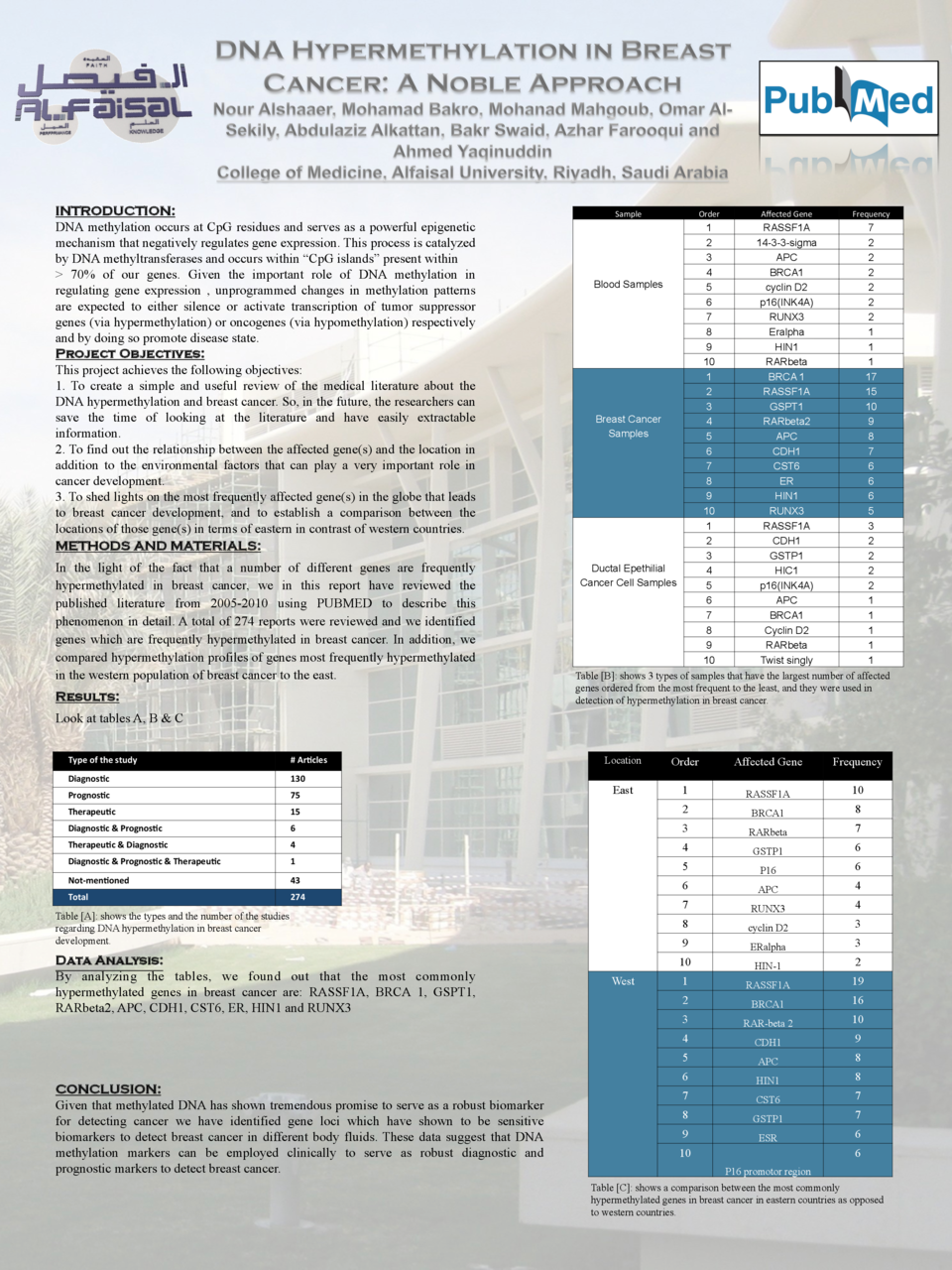
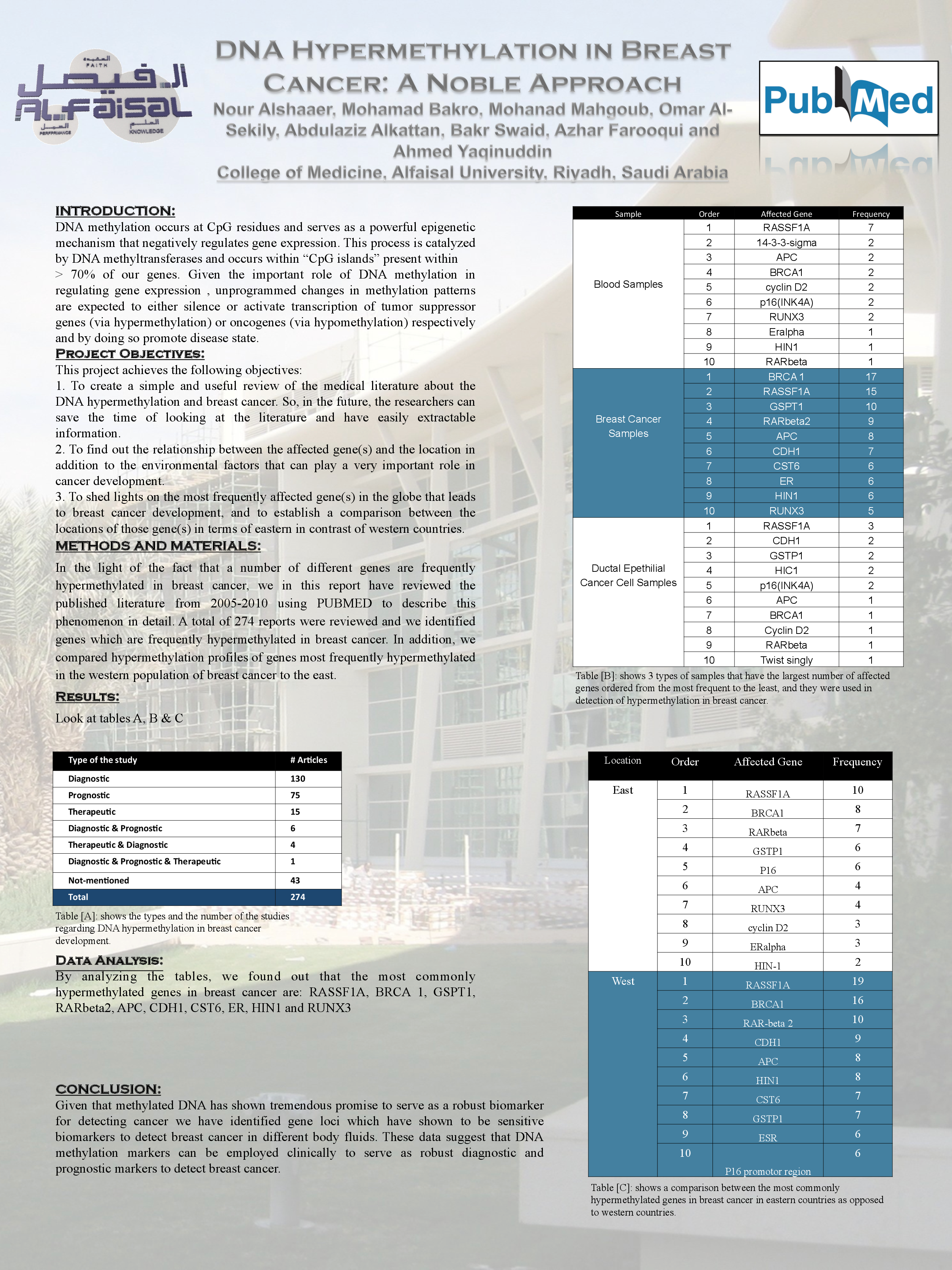
DNA methylation occurs at CpG residues and serves as a powerful epigenetic mechanism that negatively regulates gene expression. This process is catalyzed by DNA methyltransferases and occurs within “CpG islands” present within > 70% of our genes. Given the important role of DNA methylation in regulating gene expression , unprogrammed changes in methylation patterns are expected to either silence or activate transcription of tumor suppressor genes (via hypermethylation) or oncogenes (via hypomethylation) respectively and by doing so promote disease state. In the light of the fact that a number of different genes are frequently hypermethylated in breast cancer, we in this report have reviewed the published literature from 2005-2010 using PUBMED to describe this phenomenon in detail. A total of 274 reports were reviewed and we identified genes which are frequently hypermethylated in breast cancer. In addition, we compared hypermethylation profiles of genes most frequently hypermethylated in the western population of breast cancer to the east. By analyzing the tables, we found out that the most commonly hypermethylated genes in breast cancer are: RASSF1A, BRCA 1, GSPT1, RARbeta2, APC, CDH1, CST6, ER, HIN1 and RUNX3